

癌症是典型的多细胞增殖失控性疾病。一般认为,地球生命在6亿年前出现多细胞生命,现存的海绵类多孔动物门是最早出现的多细胞动物。人类的疾病多少与先天发育不足有关,另外一些与后天的损伤有关。人体内的主要系统只有2种:代谢和免疫,所有的疾病不外此2种。
1亿年前,鼠与人类的共同祖先出现。

人与人之间的基因组相似度在99.99%。我们血缘关系最近的亲属——黑猩猩,在基因上与我们人类的相似度达到了96%。人类和猕猴共有93%的DNA。猫的基因与我们人类有着90%的相似性。
当涉及到蛋白编码基因时,老鼠与我们有着85%的相似度,但是在非编码基因上,只有大约50%的相似度。国家人类基因研究所把这种相似性归咎于大约8千万年前的共同祖先。几乎所有的人类基因在老鼠身上都有个明确的对应基因,但是蛋白质编码基因只占据了各自基因组的1.5%,基于此,能治愈老鼠的化合药物通常对人类无效。
2009年《科学》杂志上的一篇报道,家牛与我们人类共享大约80%的基因。鸡与人类共享了大约60%的基因。果蝇与人类共享了61%的致病基因,这对于美国宇航局探索太空旅行可能对人类基因产生的影响非常重要。香蕉也与人类共享了大约60%的基因。
目录
一、免疫发展史
二、免疫学原理
三、主要抗癌免疫疗法
四、免疫状态评估
五、免疫治疗毒性和疗效评估
六、ICIs耐药现象的解释
七、临床应用
一、免疫治疗简史
免疫系统是多细胞动物特化的一个系统,用以维持多细胞动物在细胞层面和分子层面的稳定性。在单细胞动物中存在的原始基本机制是多细胞化后复杂机制的基础,类如在单细胞动物中的分子系统CRISPR-cas系统就是一个维护单细胞稳定性的免疫机制。

1909年Paul Ehrlich提出免疫监视学说,认为免疫系统可以遏制肿瘤的发生,免疫功能异常是肿瘤发生的基本原因之一。
1959年Frank Macfarlane Burnet和Lewis Thomas(曾任MSK院长)提出了“免疫监视(tumor immune surveillance)”的假说,该假说认为免疫系统能够识别并清除恶性肿瘤,从而抑制了肿瘤的发生发展。
2002年Gavin P Dunn和RobertD Schreiber等首次提出了肿瘤免疫编辑(Tumor Immunoediting)学说,系统阐述了癌症和免疫系统之间的三阶段关系。

二、抗癌免疫学的基本原理
100余年人类对免疫系统的不懈研究,目前已经对该领域的主要问题有了一些粗浅的认识。陈列平教授将人类对抗癌免疫的认识过程总结为,三大基本问题:有没有?免疫原性的状态如何?启动抗癌免疫的相关因素。即:
(一)人类机体内是否存在清除癌细胞的免疫反应?
答案是肯定的!
1868年一位叫威廉.布什(WilhelmBusch)的医生第一次报道,有意使用丹毒感染癌症病人后肿瘤显著缩小。1891年,美国纽约纪念医院骨科医师威廉.科利(WilliamB.Coley,1862~1936)开始以注射细菌进入肿瘤的方法治疗癌症,创立“科利毒素”疗法。这种方法疗效并不稳定,而且可能死于感染;改进后混合加热过的细菌液变得安全,经这种方法确实有不少人恶性肿瘤在无药可医的情况下得到了缓解,甚至是长期缓解。曾经一睹被打压的早期免疫治疗探索今天已经得到世人的认可,并以其名字命名了免疫界最高奖项为威廉.科利奖。
如细胞因子疗法、过继免疫细胞疗法及免疫检查点抑制剂疗法的效果已经被证实,其中有广泛意义的是ICIs,在多数癌种均有一定比例的患者获得疗效,和既往的化疗及分子靶向治疗的机理完全不同。
(二)是否存在特异性的抗原以引发清除癌细胞的特异性免疫反应?
癌细胞来源与人体自身细胞,存在抗原,但是经历了免疫编辑后的癌细胞抗原性较弱,难以有效激发免疫反应。而且在肿瘤微环境局部存在复杂的免疫抑制或免疫逃逸,导致免疫反应不能有效完成。
抗原是激发免疫反应的起点,未来这个问题尚需要继续充分研究。比利时的Thierry Boon教授长期研究癌细胞抗原问题。对于一个已经进入临床阶段并进展的癌症病灶(原位持续增殖或者可转移的),有没有癌细胞独有的抗原?进展状态的病灶存不存在人体细胞在任何时期都没有表达过的抗原性质(免疫原性)的物质?
目前看癌症抗原领域有如下观点:
癌细胞存在独有的抗原吗?还是共享的?例如有些CEA/AFP是人类胎儿时期的蛋白质,且功能现在也不清楚。癌变后,这些本来应该沉没的基因重新激活并大量表达。不知道癌细胞是紊乱表达还是功能性表达。存在癌细胞专有表达的基因吗?目前看是有的,很少,要么表达少,要么已经被免疫掉,要么免疫原性不足。
比利时学者Thierry Boon长期研究抗癌免疫反应的核心问题癌症抗原,他说癌细胞的抗原性才是主要问题。抗原性不足是当前TME免疫抑制/逃逸的解除只能解决部分问题的一个重要原因。

BOX:癌症抗原问题:没有抗原性就没有免疫反应
抗原本身的因素
异物性
抗原与机体的种系关系越远,其差异越大,免疫原性也就越强。
1)异种间的物质:病原微生物、动物免疫血清对人是良好抗原。
2)同种异体间的物质:人红细胞表面ABO血型抗原系统及同种异体皮肤和器官上的组织相容性抗原。
3)自身抗原:自身物质一般无抗原性。
a:与淋巴细胞从未接触过的自身物质(如晶状体蛋白)
b:自身物质理化性状发生改变(外伤、感染、药物、电离辐射等)
理化性状
1)分子大小
一般说来分子量越大,抗原性越强。具有抗原性的物质,分子量一般在10.0kD以上,个别超过100.0kD,低于4.0kD者一般不具有抗原性。
2)化学结构的复杂性,
蛋白质,芳香族氨基酸为主者,尤其是含酪氨酸的蛋白质,抗原性强,非芳香族氨基酸为主者,抗原性较弱。
3)分子构象和易接近性
4)物理状态,
一般聚合状态的蛋白质较其单体免疫原性强,颗粒性抗原强于可溶性抗原。
免疫途径和抗原剂量
具备上述条件的抗原物质可因进入机体的途径和剂量的不同而免疫效果迥异。
人工免疫时,多数抗原是非经口进入(皮内、皮下、肌肉、静脉、腹腔注射)机体才具有抗原性。
机体方面的有关因素
1)宿主与抗原来源的种系进化关系
2)宿主的遗传背景
3)机体的健康和营养状况
以上几方面因素在一定程度上是相互制约的。
(三)如何激发抗癌免疫反应?
激发免疫反应涉及到很多复杂的方面,如果有时间精力也可以做一个像Daniel Chen的框架图,就现状而言,我们仍处在暗室之中,只是偶尔发现了几个分子开关,更加复杂的网络还没有被发现。
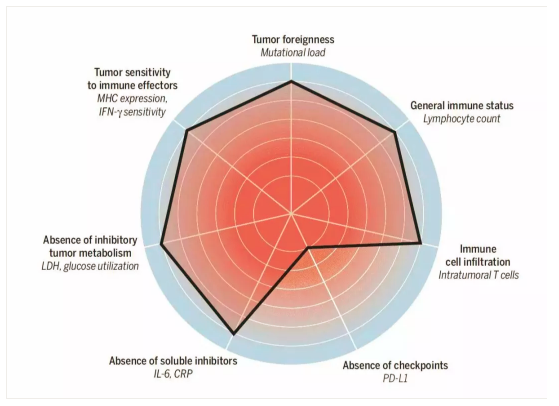
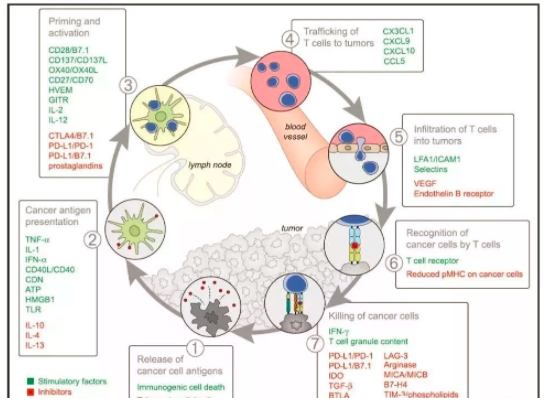
荷兰Christian U Blank在科学杂志撰文描述了影响抗癌免疫反应的7大因素,并用雷达图描绘出来。大概可分类为几类:
① 癌细胞本身的:如有氧酵解水平的LDH,细胞膜表面的抗原负荷、MHC表达,负反馈作用的PD-L1表达。
② 血液中的:CRP、IL-6所反应的慢性炎症水平;这些标志物反映了人体内有不能清除的抗原异物。一如生理性自身免疫反应在于清除体内衰老的废物代谢物一样,使用的是IgM快速清除机制,而如果是病理性自身免疫反应就需要动用慢性炎症反应机制,IgG系统。还有一个是循环血液中的免疫细胞数,这个反应了宿主的免疫系统状态。
③微环境中的:TME中的TIL反应了免疫细胞是否能够进入最后阶段,这一阶段停止于局部的免疫抑制或免疫逃逸。
值得注意的是Blank没有讨论微生态在抗癌免疫中的价值,或许是缺乏可检测标准吧。微生态是一个重要的方向,科学杂志的癌症免疫治疗专辑曾专门发文讨论。
三、主要抗癌免疫疗法
抗癌免疫疗法的历史悠久,最早是1868年一位叫威廉.布什(Wilhelm Busch)的医生第一次报道,有意使用丹毒感染癌症病人后肿瘤显著缩小。丹毒(erysipelas)是一种累及真皮浅层淋巴管的感染,主要致病菌为A组β溶血性链球菌。诱发因素为手术伤口或鼻孔、外耳道、耳垂下方、肛门、阴茎和趾间的裂隙。
1891年,美国纽约纪念医院骨科医师威廉.科利(William B.Coley,1862~1936)开始以注射细菌进入肿瘤的方法治疗癌症,创立“科利毒素”疗法。这种方法疗效并不稳定,而且可能死于感染;改进后混合加热过的细菌液变得安全,经这种方法确实有不少人恶性肿瘤在无药可医的情况下得到了缓解,甚至是长期缓解。
由于20世纪初更加安全有效的放射疗法已经开始普及,科利毒素疗法开始被MSK医院禁止,直至后来被ASCO禁止,虽然ASCO最终撤销了禁令。后来科利的女儿继承了父亲的遗志,曾出巨资资助癌症免疫治疗研究。曾经一睹被打压的早期免疫治疗探索今天已经得到世人的认可,并以其名字命名了免疫界最高奖项为威廉.科利奖。
根据已知的免疫学原理,推测这种利用细菌感染治疗癌症的机制是以细菌抗原调高机体对肿瘤的反应性,是先天免疫和后天免疫均处于激活状态。细菌和自身衰老损伤细胞即癌变细胞均为同一种免疫机制清除,之间并无严格的界限,甚至有相当部分功能处于重叠状态。
然而由于人类对自身小宇宙中的这个神秘系统认知的局限性,很长时间以来人类并没有找到可靠的免疫治疗方法。直到两种免疫治疗手段出现:
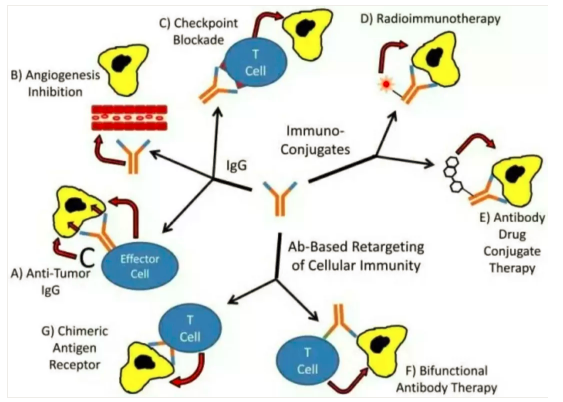

1. 针对癌细胞的抗体治疗
癌细胞上有一些特殊的抗原,人类使用免疫学抗体原理清除他们以达到治疗癌症的目的,这些治疗取得了一定疗效。
赫赛汀单抗:针对癌细胞的生长刺激因子,即人类表皮生长因子受体2(human epidermal growth factor receptor-2,HER2)。该基因,即c-erbB-2基因,定位于染色体17q12-21.32上,编码相对分子质量为185000的跨膜受体样蛋白,具有酪氨酸激酶活性。这个受体被阻滞后,癌细胞的生长转移特性受到抑制。但是由于癌细胞不止一种生长刺激因子,所以即使癌细胞初始表达这种受体,但是初始仅有50%左右的病人对这种治疗有效,几乎所有病人会在一年内对该抗体治疗耐药。
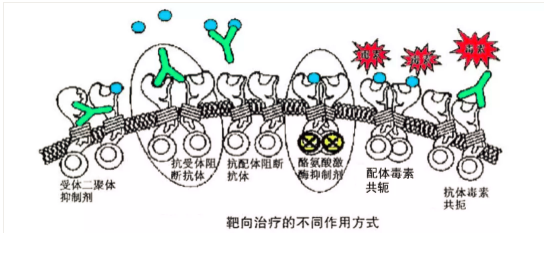
2. 细胞间质如血管是癌症病灶必须的,针对癌症病灶中的细胞间质治疗。
AVASTIN(贝伐单抗):针对VEGF这种细胞因子的抗体,可以引发抗体反应清除这种细胞因子,阻断体内新生血管的形成来阻滞癌症的进展,代价是人体正常的血管新生的生理过程也受到干扰。
3. 免疫检测点抑制剂(ICIs)
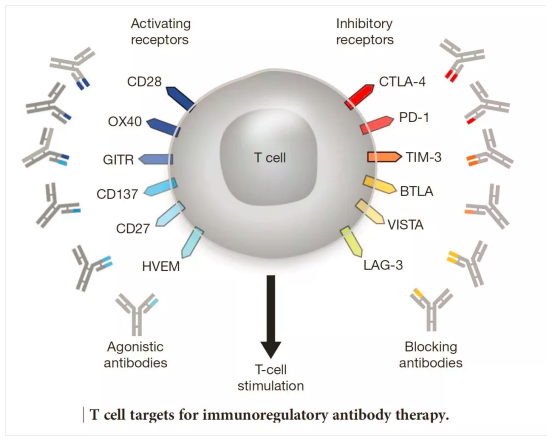
免疫检测点是指位于效应T细胞上的一些激活性和抑制性受体调节开关,使用激活性抗体可以启动该受体下游的功能(激活或者抑制),激活可以使得T细胞处于攻击状态,抑制可以使得T细胞处于安静状态。人体内有100万亿个种类各异的真核细胞,如何精准地调节T细胞的免疫攻击性能是一个受到多种机制调节的过程,其中免疫检测点是其中的一种调节机制。
第一个被应用于医疗实践的免疫检测点是CTLA-4,由美国学者James Allison发现并花了10年左右证实其功能。第二个被发现的该类分子是PD1/PD-L1,PD1首先被日本学者本庶佑发现,并被证实其效用;而PD-L1是被美籍华人学者陈列平发现的,后也被证实其效用。
当前的主要ICIs就是基于这几个靶点设计的。免疫检测点由很多,其它分子也有潜在的开发价值,相关药物正在研发中。
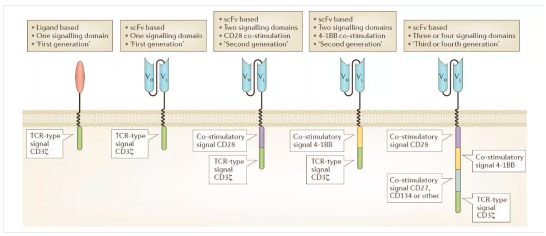
4. 过继细胞疗法CAR-T细胞疗法
过继细胞治疗经历了漫长的探索过程,早期的过继细胞治疗一般是指:第1代LAK细胞。第2代CIK细胞,第3带TIL细胞,第4代抗原特异性的CTL细胞。
早期的过继细胞疗法有效率低下,而且制备过程难以标准化评估,已经被基本淘汰。目前流行的是第5代过继细胞治疗法,技术有2种,除了CAR-T细胞,还有一种叫TCR-T细胞。
四、免疫评估
ICIs制剂的疗效主要和以下几个因素相关:
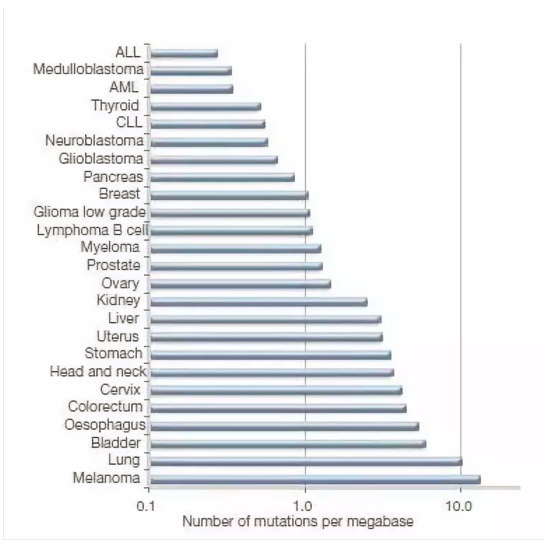
1. 新抗原数量:突变负荷是指正常等位基因(A)突变成有害等位基因(a)形成在选择上不利的纯合体(aa)所引起群体适应度下降的现象。如:人类的一些代谢遗传病,象黑尿症、半乳糖血症等。
用肿瘤突变负荷(TMB)来定量:被定义为每百万碱基中被检测出的,体细胞基因编码错误、碱基替换、基因插入或缺失错误的总数。具体操作就是测量某种肿瘤体细胞内编码蛋白的碱基突变数量,包括替换、插入/缺失等各种形式的突变;
量化的描述TMB,可分为4类:
1)TMB-High=>20mutations/Mb;
2)TMB-Intermediate, 6-19 Muts/Mb;
3) TMB-Low <= 5 Muts/Mb;
体细胞突变可能受到外源性诱变因素的影响,比如肺癌中烟草(吸烟)诱导的C→A的突变。恶性黑色素瘤中,紫外线照射引起的C→T的突变。内源性因素引起的突变可以是DNA错配修复突变,比如结直肠癌和食管癌中的MSI(微卫星不稳定)。在实体肿瘤中,95%的突变为单个碱基的替换,造成的非同义突变(一个核苷酸突变改变一个蛋白的氨基酸顺序),错义突变(非同义点突变,单个核苷酸改变导致一个密码子编码一个不同的氨基酸)和无义突变(非同义点突变使一个密码子变为终止密码子引起多肽链合成提前终止),共同构成了体细胞非同义突变的基本要素。
2. TMB相关因素:肿瘤突变负荷(tumormutation burden,TMB)、微卫星高度不稳定(microsatellite instability-high, MSI-H)和错配基因修复缺失(mismatch-repairdeficiency,MMR),这些潜在的bio-marker又指向一个共同点突变相关新抗原(mutation-associated neoantigen)。
3. 肿瘤微环境种的PD-L1表达丰度及TIL数量。具体见荷兰Christian U Blank在科学杂志撰文并画出的影响抗癌免疫反应的7大因素雷达图。
4. 微生态:《科学》杂志发表的五篇肠道微生物影响肿瘤免疫治疗的研究文章,来阐述免疫疗法疗效与肠道微生物的关系。
2015年11月27日,《科学》杂志发表了两篇研究,首次指出癌症患者肠道微生物的组成,决定了以检查点抑制剂(CTLA-4、PD-L1)为代表的癌症免疫疗法的有效性。由法国GustaveRoussy 癌症中心的免疫学家Laurence Zivogel博士领导的研究小组发现,肠道中具有多形拟杆菌和脆弱类杆菌的小鼠,在接受CTLA-4抗体治疗时,会表现出更好的治疗效果;而无菌小鼠的肿瘤则对该治疗几乎没有响应;在给无菌小鼠移植特定菌株后,可以增强CTLA-4抗体疗法的有效性。来自芝加哥大学的Thomas Gajewski团队,也通过类似的方法,证明了双歧杆菌属有利于PD-L1抑制剂的抗肿瘤效果。
2017年11月2日,《科学》杂志再次同时在线发表了两篇重磅论文。
一篇《Gut microbiomeinfluences efficacy of PD-1–based immunotherapy against epithelial tumors》还是由两年前的Laurence Zivogel博士团队研究撰写,这次的研究对象从老鼠转移到了人类。该团队对接受过PD-1抑制剂治疗的249例癌症患者进行了分析,其中69例患者因常规原因(如牙科治疗或尿路感染),在接受PD-1治疗前后服用了抗生素。那些服用抗生素的癌症患者,因为抗生素导致肠道菌群紊乱,在接受PD-1抑制剂治疗之后,癌症很快就复发,总体生存期(OS)竟然比未服用抗生素的患者缩短了近45%!
研究人员还将人类患者的肠道微生物移植到无菌小鼠身上,结果发现移植那些治疗有效患者肠道微生物的小鼠,接受PD-1/L1抑制剂治疗同样会有效,而移植无效患者的肠道微生物,治疗也就无效。具体试验过程如下:
①分析249名患肺癌、肾癌等肿瘤并接受抗PD-1免疫抑制剂治疗的患者,69名同时接受广谱抗生素(ATB);
②ATB患者平均总生存期为8.3个月,远远低于非ATB患者的15.3个月;
③患者体内Akkermansia muciniphila(Akk菌)的相对丰度与对免疫检查点抑制剂(ICI)的响应显著相关;
④将对ICI响应的患者的粪菌移植给无菌小鼠,可以改善PD-1抑制剂对小鼠肿瘤的效果;
⑤移植对ICI不响应的患者粪菌的无菌小鼠,在口服Akk菌后,也能恢复对PD-1抑制剂的响应。
同时刊发的另一篇论文中,来自美国德克萨斯州大学安德森癌症中心的研究人员,对112名接受PD-1抑制剂治疗的癌症患者进行了口腔和肠道微生物的分析。结果发现免疫疗法有效与无效患者的肠道菌群存在显著差异,证实肠道微生物在免疫疗法中扮演了极其关键角色。
2018年1月5日,ThomasGajewski团队登顶《科学》封面,肠道微生物影响人类癌症患者免疫治疗效果再次得到证实!研究人员通过对42名转移性黑色素瘤患者的粪菌构成进行分析,结果证明患者肠道菌群与PD-1免疫疗法的效果之间存在显著关联,并在对治疗响应明显的患者肠道菌群中,鉴定出高丰度的长双歧杆菌、产气柯林斯菌和屎肠球菌,并发现在给无菌小鼠移植这些人类患者的肠道微生物后,能够显著增强小鼠的肿瘤免疫应答和肿瘤控制。
有人会问,为什么不同的研究团队鉴定出的菌群种类不完全一致,North Carolina大学Scott Bultman教授表示:“研究对象所处地理位置和饮食习惯不同,肠道菌群不同是正常的,而且不同研究之间也存在一定的重合度。”
另外一些因素:
疗效好的:吸烟、近期放疗
疗效差的:EGFR基因突变。
KRAS突变,似乎不影响疗效:总体人群的临床获益率为29%,携带KRAS突变的患者临床获益率是36%,两者无统计学差异。关于这一点,要做一下额外的补充:事实上,KRAS突变的非小细胞肺癌可以分为三类。
(1)同时携带KRAS和P53突变的患者,这类患者对PD-1治疗敏感;
(2)同时携带KRAS和STK11突变的患者,这类患者对PD-1治疗抵抗;
(3)其他患者,对PD-1治疗的有效率居中。
在本次研究中,①携带STK11突变的患者,接受PD-1抑制剂治疗,的确是有效率更差的;②JAK突变、B2M突变的患者,要当心,③PD-L1表达和TMB高低无关④MDM2、MDM4扩增的患者,是否会发生超进展,有待进一步研究。
五、毒性管理与疗效评价
细胞因子释放综合症(Cytokine Release Syndrome, CRS):
在CAR-T细胞治疗过程中发生的一种过度免疫反应。临床表现主要包括:发热;特征性细胞因子升高;和临床毒性的存在(表1)。并发CRS患者的典型发烧,通常在回输CAR-T细胞后约24小时开始并且可以持续数天。然而,发烧并不总能够预示临床发生相关毒性的多少,严重程度和发病趋势。
至少7个细胞因子水平在血清中的增高与患者发生CRS具有明确的相关性,这些细胞因子是:IFN-γ (干扰素-γ),Fracktalkine (分形趋化因子),GM-CSF (粒-巨噬细胞生长因子),IL-5(白细胞介素-5),IL-6(白细胞介素-6),Flt-3L (人FMS样酪氨酸激酶3配体)和IL-10 (白细胞介素-10)。
7种细胞因子在需要治疗的重症CRS患者中,至少有2项比治疗前基线水平增高75倍。重症CRS患者都出现了至少一个下述临床表现:缺氧,低血压,和/或神经系统的异常改变。
因此,结合临床和实验室数据,可以根据以下3项指标来确诊患者CRS的发生:
1. 持续发烧超过3天(~38C°);
2. 选择性细胞因子升高;
3.伴有临床毒性反应的证据。
上述标准可以将患者分为重症CRS组和非CRS组。非CRS组患者包括未发生CRS患者,也包括仅有低热和轻度细胞因子增高的轻度CRS患者。重症CRS患者需要密切观察和治疗干预。非CRS患者仅需要常规观察和一般性对症处理。重症CRS患者平均住院时间为56.7天(SD=28.6;范围20-104天)。非CRS患者平均住院15.1天(SD=18.8;范围:4-61天)。
尽管在CAR-T治疗引起的严重副作用发生之前,检测血清CRS相关细胞因子,可以指导临床诊断和治疗,但是每天快速实时检测细胞因子,因技术上的限制并不现实可行。研究发现,血清急性反应蛋白之一,C-反应蛋白(CRP)水平的增减与CRS时患者血清IL-6水平和IL-6受体阻滞剂治疗后显著相关。同样C-反应蛋白水平与类固醇药物对CRS的疗效显示了明显的负相关。因此认为C-反应蛋白是预测CRS的一个良好指标。C-反应蛋白水平超过正常阈值预示具有CRS发生的高度危险性,C-反应蛋白测定的敏感性约86%,而特异性为100%。

细胞因子释放综合症的处理
重症CRS产生的不良反应和毒性作用需要有效和强力的医疗干预。通常包括血管活性升压药支持,呼吸机支持,使用抗癫痫药和解热镇痛药物。尽管这些毒副作用令人担心,迄今为止,在正确有效的医疗处理条件下,临床上CRS已经是完全可逆性的。
采用大剂量类固醇激素相当于每天100mg以上强的松剂量,可以迅速逆转CRS的临床症状。然而,问题的另一方面是,类固醇药物也能抑制CAR-T的体内增殖,使得使用类固醇类药物治疗CRS的患者,有较高的复发率,影响CAR-T治疗的疗效。
白细胞介素-6受体(IL-6R)阻断性单克隆抗体药物(Tocilizumab)也可以用来治疗和改善CRS的毒副作用。已有临床试验证明,阻断IL-6受体后能迅速解决CRS带来的毒副作用。在3例CRS患者中(患者血清中IL-6浓度增高达正常浓度的27-400倍),单独使用Tocilizumab抗体后,患者发烧和CRS症状在1-3日内明显缓解,疗效与类固醇疗法相似,外周血检测显示Tocilizumab治疗对CAR-T细胞体内增殖没有影响。
CAR-T治疗能引起对神经系统的毒性作用。患者可能出现可逆性神经系统并发症。包括神志不清和癫痫样症状。患者可以发生渐进性的神志混乱,词语困难,失语,可最终发展到反应迟钝。有些患者的神经系统并发症发展到需要气管插管及机械辅助通气等措施,以便对呼吸道加以保护。对神经系统并发症的检查,CT和磁共振成像常无特别发现。脑电图能确诊癫痫样脑电波活动,有助于指导抗癫痫治疗的处理。
ICIs治疗相关毒性管理:
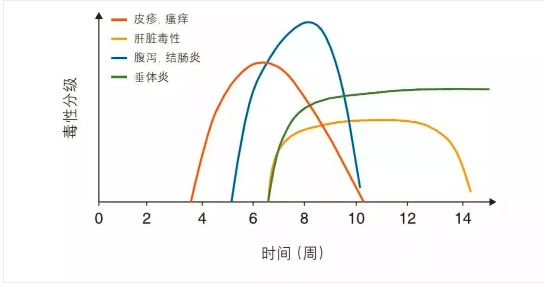
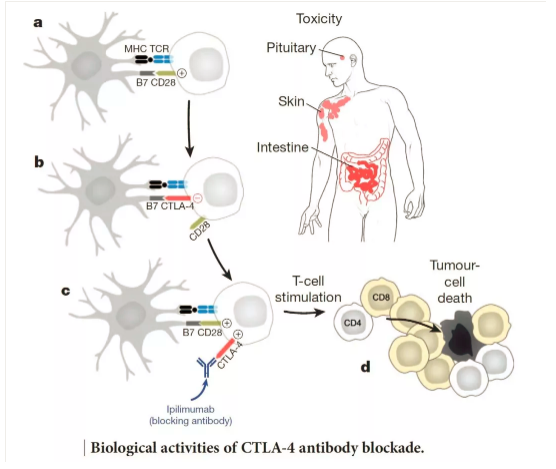
针对细胞毒T淋巴细胞相关抗原4(cytotoxicT lymphocyte-associated antigen 4, CTLA4)和程序性死亡受体(programmed death-1 receptor,PD-1)及其配体PD-L1的单克隆抗体(monoclonalantibodies,MoAbs)治疗已经成为越来越多肿瘤的标准治疗。越来越多的患者应用这些药物的同时也可能会出现治疗相关的毒性。毒性发生率在不同的免疫检查点抑制药物之间也不尽相同。
免疫检查点抑制剂(immunecheckpoint inhibitors,ICPi)的毒性可以分为输注反应和免疫相关不良事件(immune-related adverse events,irAE)或特殊关注的不良事件(adverseevents of special interest,AEoSI)。ESMO临床实践指南仅涉及后者。虽然某些脏器的irAE更加常见,但实际任何器官和组织都有可能受累。最常发生的irAE主要累及皮肤、结肠、内分泌器官、肝脏和肺;其他组织和器官虽然少见,但有可能相对更严重、甚至是致命的,比如神经系统病变和心肌炎。
Ipilimumab的免疫相关毒性
Ipilimumab,一种抗CTLA4单抗,临床剂量为3mg/kg时,60%~85%的人群出现irAE:大多数是1~2级毒性,约10%~27%的人会发生3~4级毒性,在早期的3期临床研究中有2.1%的患者出现ipilimumab相关的死亡。这些毒性发生的时间各不相同,但大多出现在治疗开始后的8~12周[图中列出了ipilimumab治疗后不良事件(adverseevents,AE)的发生时间],皮肤毒性常为首发症状。这些毒性特征表现为剂量依赖性:ipilimumab剂量为0.3 mg/kg时并没有观察到3~4级的不良事件,而使用10mg/kg时,3~4级毒性上升到30%。当ipilimumab 10 mg/kg继以维持剂量作为辅助治疗时,3~4级irAE的发生率为41.6%,5级irAE的发生率为1.1%。对于1-2级的皮肤AE,继续应用(至少1周)ICPi。如果出现瘙痒,则开始应用外用润肤剂、抗组胺药和/或糖皮质激素(低强度)乳膏。当AE≤1级时重新开始应用ICPi。对于3级的皮肤AE,暂停ICPi,并立即应用外用润肤剂、抗组胺药和高强度的糖皮质激素乳膏[II, B]。对于4级的皮肤AE,停用ICPi(永久),考虑收患者入院,并立即与皮肤科医生会诊。开始应用经静脉给药的糖皮质激素[1–2 mg/kg(甲基)强的松],并根据AE的缓解情况逐渐减量[II, B]。
疗效评价
目前的试验结果发现肿瘤微环境中PD-L1和淋巴细胞双阳性的病人在黑色素瘤中有45%,肺癌中有17%, 膀胱癌中有32%,这个比例和临床试验抗PD-1/PD-L1治疗的反应率很相似。
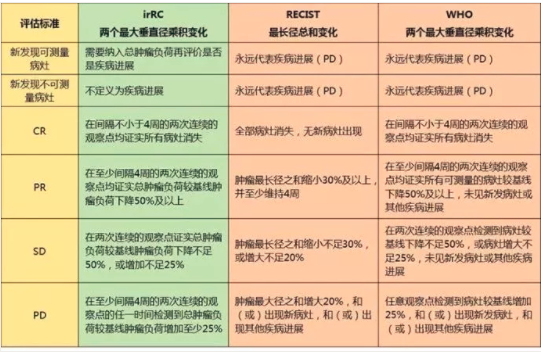
2009年国际上建议并制定肿瘤免疫治疗的疗效判断新标准—免疫相关反应标准(Immune-RelatedResponse Criteria,irRC),以弥补RECIST或改良后的WHO标准并不完全适用于抗肿瘤细胞免疫治疗的缺陷。
六、ICIs耐药现象的解释
为何有些肿瘤抵抗PD-1/PD-L1治疗,陈列平教授将耐药性肿瘤分为3类:
① Target-missingresistance:这些肿瘤不表达PD-L1,或者有PD-L1但没有T淋巴细胞(缺乏PD-1),因此属于靶子缺失型。这种肿瘤对PD-1/PD-L1抗体治疗不可能有效果。明显有其他逃逸机制在起作用。
② Primaryresistance:肿瘤微环境中有PD-L1,也有T细胞,但病人对PD-1/PD-L1抗体治疗没有反应。这样的情况比较少见。
③ Acquiredresistance:肿瘤微环境中有PD-L1,也有T细胞。病人对PD-1/PD-L1抗体治疗最初也有反应,但是后来肿瘤复发,用PD-1/PD-L1抗体再治疗也无效。这一种才是真正的耐药。
MD安德森的Padmanee Sharma发表文章《Primary, Adaptive, andAcquired Resistance to Cancer Immunotherapy》讨论ICIs的耐药机制。
肿瘤免疫疗法的一个显著特点是肿瘤响应的时间长,但是在使用anti-CTLA-4或anti-PD-1治疗黑色素瘤的患者中,大约有1/4-1/3的患者在经历初始响应后出现了肿瘤进展,即获得性耐药。获得性耐药的产生的主要是肿瘤细胞上特定基因或通路的表达或上调,从而导致肿瘤微环境中免疫细胞的浸润以及功能受到抑制。例如,T细胞功能的丧失,肿瘤抗原提呈出现缺陷以及其他一些新突变的产生。例如,在一类较晚获得anti-PD-1耐药性的病人中,发现了B2M的突变,从而导致抗原提呈机制缺陷。在另两例获得性耐药肿瘤中,发现了IFNγ通路中JAK1或JAK2发生了突变。PD-1抗体只是解除了“T细胞枷锁”,疾病的控制最终还是要通过T细胞实现,因此,凡是影响T细胞招募、激活等功能的,均能够最终影响PD-1抗体的临床收益。
导致免疫疗法耐药的肿瘤内在原因:
①MAPK通路的激活与或PTEN表达的缺失而引起的PI3K通路的增强:
癌基因信号通过MAPK通路导致VEGF与IL-8的产生,从而抑制T细胞的招募与功能。肿瘤抑制基因PTEN表达缺失从而PI3K通路增强,这与IFNγ,颗粒酶B的基因表达量降低以及肿瘤浸润CD8+T细胞的数目减少是高度相关的。
②WNT/β-catenin信号通路的持续表达:
癌基因信号通过稳定β-catenin导致WNT信号通路持续激活,从而将T细胞排除在肿瘤之外。在人Non-T-cell-inflamed的黑色素瘤中,肿瘤内在的β-catenin信号基因高度表达,且在肿瘤微环境中缺少T细胞与CD103+DC细胞。
③肿瘤上PD-L1的高表达:
肿瘤上高表达的某些配体如PD-L1,会抑制抗肿瘤T细胞的应答。多种机制可能导致PD-L1高表达,包括PTEN的缺失或PI3K/AKT的突变,EGFR突变,MYC过表达,CDK5基因破坏以及PD-L1基因3’-UTR的截短。目前还不知道PD-L1的高表达是否会对anti-PD-1/PD-L1的响应有影响,但是它会影响其他的肿瘤免疫疗法。
IFNγ信号通路的缺失:由肿瘤特异的T细胞产生的IFNγ,能够识别肿瘤细胞或抗原递呈细胞上的相应受体,从而发挥有效的抗肿瘤免疫响应。IFNγ能够增强MHC分子的表达,从而增强肿瘤抗原提呈作用。IFNγ也能够招募其他的免疫细胞,或者直接抑制肿瘤细胞的增殖,促进其凋亡。因此肿瘤细胞上IFNγ通路相关蛋白,如IFNγ受体IFNGR1与IFNGR2,IFNγ受体链JAK1与JAK2,STATs,IRF1等突变与缺失,都会导致对免疫检查点抑制剂的耐药。
④缺少肿瘤抗原:
免疫疗法依赖于肿瘤抗原特异的T细胞。在人黑色素瘤,肾细胞癌,非小细胞肺癌中,DNA突变频率高,肿瘤免疫原性更强,因而对anti-PD-1疗法响应更好。而在胰腺癌以及前列腺癌中,DNA突变频率低,肿瘤免疫原性低,对anti-PD-1疗法响应差。
⑤抗原提呈机制存在缺陷:
在某些情况下,由于抗原加工过程中的蛋白酶体成员,转运蛋白,MHC本身以及beta-2-微球蛋白(B2M)的功能缺陷,会导致抗原提呈机制不能有效地将肿瘤抗原提呈到细胞表面。B2M在HLAI家族的折叠与转运到细胞膜的过程中发挥关键作用,若其丧失功能,则CD8+T细胞失去了识别功能。
⑥存在一系列特定基因的表达:
在对PD-1疗法没有响应的肿瘤中,有一些基因表达被富集,被称为innate anti-PD-1 resistance signature,或IPRES。这些基因与间叶细胞的转化,全能型以及伤口愈合相关,且更倾向于表达在胰腺癌等对PD-1不响应的肿瘤中。
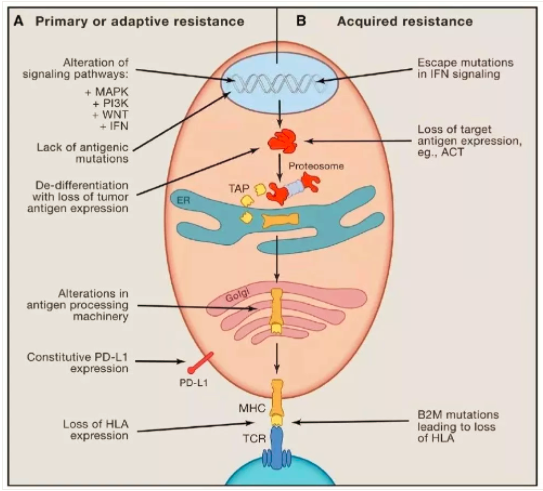
导致原发性/继发性耐药的肿瘤外部原因:
这些原因是由于肿瘤微环境中一些成员发挥抗癌免疫响应的抑制作用,主要包括调节T细胞Tregs,髓样抑制细胞MDSCs,M2巨噬细胞,其他的抑制性免疫检查点与抑制性细胞因子等。
① Tregs:
Tregs能通过分泌抑制性细胞因子或者通过直接的细胞接触来抑制效应T细胞Teffs的响应。许多人肿瘤中发现了浸润的Tregs,且鼠模型中去除肿瘤微环境中的Tregs能够显著提高免疫响应。由于CTLA-4在Tregs上高表达,anti-CTLA-4能够显著提高Teffs/Tregs的比例,从而提高肿瘤对免疫疗法的响应。
② Myeloid-derivedsuppressor cells (MDSCs):
MDSCs在多种病理条件包括肿瘤,发挥着免疫响应调节因子的作用。人MDSCs表达CD11b+与CD33+,但是不表达HLA-DR以及系种特异的抗原CD3,CD19与CD57。MDSCs能够促进血管生长,肿瘤侵袭与转移。肿瘤微环境中MDSCs的存在与降低的生存率以及免疫检查点抑制剂疗法的响应率相关。目前实验中使用PI3Kγ抑制剂来调节巨噬细胞功能,在老鼠模型中,PI3Kγ抑制剂与anti-PD-1联用表现出了良好的肿瘤抑制效果。
③ M2macrophages:
肿瘤相关的巨噬细胞(Tumor-Associated Macrophages,TAMs)也能够影响免疫治疗的响应。TAMs包括M1巨噬细胞和M2巨噬细胞,在大多数情况下M2巨噬细胞占TAMs的大多数。其中M1巨噬细胞能够高表达IL-12,IL-23,MHC以及B7家族分子来促进抗原提呈与Th1细胞的激活,从而发挥抗肿瘤免疫作用;而M2巨噬细胞能够分泌抑制性细胞因子IL-10与TGF-β,从而抑制免疫响应与促进肿瘤生长与转移。临床上TAMs的数目越多,肿瘤预后就越差。临床前实验使用巨噬细胞集落刺激生长因子受体1(CSF-1R)的抑制剂,能够显著减少TAMs数目,抑制肿瘤生长。CSF-1R抑制剂与anti-CTLA4或anti-PD1再加上吉西他滨联用,能够有效缓解单独anti-CTLA-4或anti-PD1不响应的鼠胰腺癌模型。
④ 其他的抑制性免疫检查点:
除了PD-1与CLTA-4,T细胞上还存在其他的抑制性免疫检查点,包括TIM-3,LAG-3,BTLA,TIGIT,和VISTA等。2016年《naturecommunication》上,有研究者发现anti-PD-1耐药性的产生与抑制性免疫检查点TIM-3的的表达量升高密切相关。在两个老鼠肿瘤模型中,耐药后与给药前相比,肿瘤浸润的T细胞而非外周血或脾脏T细胞上的TIM-3表达量显著上调,且TIM-3表达量上调的主要是那些结合了anti-PD-1抗体的T细胞。
需注意的是TIM-3表达量升高是anti-PD-1疗法特异的,因为anti-CTLA-4中并未检测到类似现象。此外,肿瘤细胞上TIM-3配体Galectin-9的表达量也显著升高。当anti-PD-1疗法出现耐药后,联用anti-TIM-3,显著提高了生存率。有意思的是,当anti-PD-1和anti-TIM-3联用被耐药,肿瘤重新进展时,那些结合了anti-PD-1和anti-TIM-3抗体的T细胞上,其他的抑制性免疫检查点如CTLA-4,LAG-3的表达量明显升高。
这说明了肿瘤浸润T细胞的抑制性免疫调节是动态变化的,存在着补偿效应。最后,在两例对anti-PD-1疗法获得性耐药的NSCLC病人肿瘤样本中,也观察到了TIM-3而非其他抑制性免疫检查点的表达量显著升高。这些结果说明了anti-PD-1和anti-TIM-3联用是对于那些anti-PD-1疗法获得性耐药病人的一个良好策略。
免疫抑制细胞因子与免疫抑制分子:
肿瘤或者巨噬细胞会释放一些免疫抑制细胞因子或者免疫抑制分子来减弱局部的抗肿瘤免疫反应。肿瘤微环境中的免疫因子或免疫细胞的异常表达
某些免疫因子如激活型免疫因子(IL-12,IL-23),抑制性免疫因子(IL-10与TGF-β),免疫细胞如Tregs, Myeloid-derivedsuppressor cells 等能够显著影响抗体的临床治疗效果。TGF-β能够促进血管生成,刺激Tregs从而发挥免疫抑制作用。
在多种肿瘤中,高水平的TGF-β都伴随着极差的预后。临床前实验使用TGF-β受体激酶抑制剂与anti-CTLA-4联用,或者放疗与TGF-β抑制剂都显示出了较好的肿瘤抑制效果。在细胞外,CD39能够将ATP水解成AMP,进一步被胞外核苷酶CD73加工为免疫抑制分子腺苷adenosine。Adenosine能够通过T细胞上的A2A受体抑制T细胞的增殖与细胞毒活性,也能通过肿瘤细胞上的A2B受体促进肿瘤转移。
多种类型的肿瘤中,CD73的高表达伴随着较差的预后,且会影响anti-PD-1的效果。此外,IFNγ会促进免疫抑制分子IDO的表达,IDO能直接负调控效应T细胞的功能。
趋化因子与趋化因子受体:
某些特异的趋化因子与趋化因子受体在MDSCs和Tregs往肿瘤微环境的运输过程中起到重要作用。肿瘤细胞能够分泌配体CCL5,CCL7和CXCL8,通过结合MDSCs上表达的CCR1及CXCR2受体,从而将MDSCs吸引至肿瘤微环境中。
CCR4在Tregs上高表达,anti-CCR4能够有效抑制T细胞的招募并通过ADCC效应减少Tregs的数目。此外CXCR4是CXCL12的受体,CXCL12能够通过影响Tregs定位等多种方式发挥免疫抑制作用。
肿瘤浸润细胞上CD28的表达:
CD28是T细胞的共刺激分子,对于T细胞的激活,增殖和存活起到关键作用。在人出生时,所有T细胞都会表达CD28,但是CD28的表达量会随着年龄的增加而下降,80岁时会有10-15%的CD4+T细胞以及50-60%的CD8+ T细胞缺失CD28表达。CD28-T细胞产生的主要原因是重复的抗原刺激。CD28表达的消失只存在与人和灵长动物中,在鼠中并没有被发现。
2017年03月09号,science期刊发表了来自Emory疫苗中心的研究工作,他们发现CD28对于耗尽的CD8+T细胞(ExhaustedCD8+ T Cell)的再激活是必须的。在老鼠模型中,阻断CD28-B7共刺激通路,会影响肿瘤特异CD8+T细胞的增殖和激活,降低对anti-PD-1/PD-L1疗法的响应。
在经过anti-PD-1治疗的NSCLC病人中,增殖的CD8+T细胞(高Ki-67表达)大都为PD-1阳性,且被激活(高HLA-DR,CD38表达)。和鼠模型中一样,这些增殖的CD8+T细胞大都是CD28阳性的,说明了CD28共刺激对于肿瘤浸润的PD-1+CD8+T细胞的增殖与再激活是至关重要的。因此,CD28可以用作预测anti-PD-1疗法响应程度的分子标记。
抗体药物失效的可能原因是巨噬细胞“吃掉”了它:
MGH研究人员将PD-1抗体opdivo、肿瘤细胞、T细胞、巨噬细胞等分别染色,发现PD-1抗体能够与巨噬细胞表面的Fcγ受体结合,从而被巨噬细胞“吃掉”,这在体外细胞实验和小鼠体内均得到证实,同时研究人员发现酶PNGaseF能过抑制该吞噬过程,这给PD-1抗体临床治疗方案的优化提供思路。
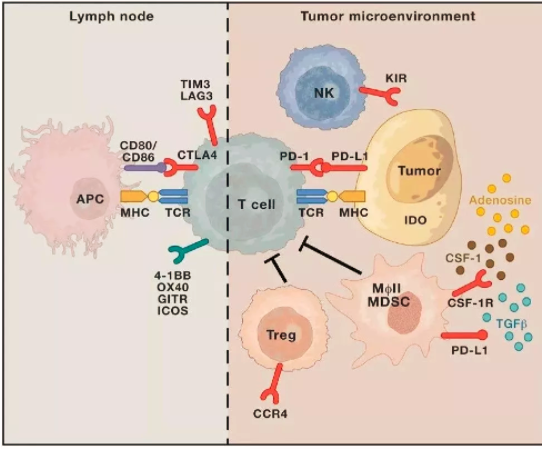
ICIs抗体药物耐药对策:
①联合用药:多个免疫检查点抑制剂药物的联合使用(opdivo联合yerovy)。
②抗体药物和化疗药物的联合使用:如keytruda+培美曲塞+卡铂联合治疗方案。
③在生物标志物的指导下采取个性化治疗方案,以获得更高的临床收益。
七、临床应用
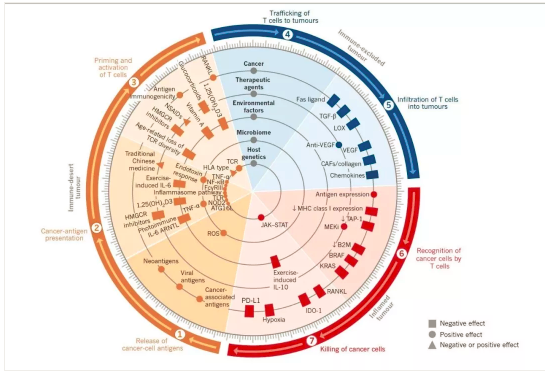
人类对癌症的认知经历了细胞与分子生物学水平的演进,目前对癌的解释可以概括为:自身体细胞在基因组突变积累的基础上出现失控增殖表型和迁移失控表型并可能表达异常信号分子,通过占位效应和分子毒性效应导致原宿主多细胞系统代谢奔溃的生命现象。
这种现象见于各种动物,但不同物种发病率不同,在人类60岁以后癌症是常见致死致残性疾病,如果一个人活到80岁有50%可能性出现癌症类现象,癌症的异质性很大,有些即使很大也未必致死,有些即使转移也能较长时间存活。
人类在癌症免疫疗法取得突破性进展之前主要的局部治疗有:外科式手术刀或者物理化学消融性微创外科、放射线治疗、化学药物、分子靶向药物(各种抗体或者小分子酪氨酸激酶抑制剂)。
当前比较成功的抗癌免疫治疗方法是CART-T过继免疫治疗和免疫检测点抑制剂。前者在血液肿瘤取得了很大的成功,后者在血液肿瘤及实体肿瘤均显示了疗效。传统的抗癌疗法与免疫治疗具有协同效应,而且传统的抗癌疗法也对免疫系统或免疫反应有一定影响:
① 化疗药物吉西他滨、卡培他滨及环磷酰胺在破坏癌细胞的同时释放的癌细胞坏死物比其它化疗或分子靶向内分泌药物更具有抗原性。
② 物理如放射线或者物理化学消融如热冷及酒精等消融技术杀死的癌细胞裂解物也具有一定免疫原性,甚至可导致残余肿瘤消退。
③ 外科大手术创伤大具有免疫抑制效应,切除过程中的癌细胞破碎可导致癌细胞脱落转移,当然也可能会导致免疫原性反应。
未来的方向是:
①了解原发和继发耐药的机制。
原发耐药:微环境中缺乏靶点,或者存在另外的免疫抑制机制。
继发耐药:TME发展出一些针对ICIs的新机制,如直接吞噬抗体。
②了解TME免疫抑制的整体框架。
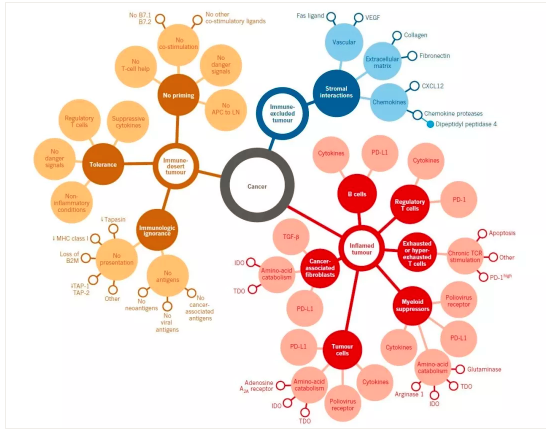
现在看来,在癌症病灶中肯定存在着一种类似保护罩似的局部结构,我觉得可以从以下几方面理解:
-
全身性的:癌症细胞群可以释放全身性的代谢和免疫抑制剂,以调节全身全身的代谢与营养、免疫状态。
-
局部可溶性分子(来自癌巢中的癌细胞和间质细胞):在局部微环境形成高浓度的抑制溶剂。
-
局部不可溶解性的(来自癌巢中的癌细胞和间质细胞):间质里的胶原蛋白纤维间隔等组织骨架。其实感觉组织骨架就是植物细胞的细胞壁似的物质,或者类似于植物的纤维素,构建组织微环境并支持多细胞系统的基本形态。
-
癌细胞表面的:典型的如PD-L1。
-
间质细胞层次的:如MDSC、癌性相关纤维母细胞和癌性巨噬细胞。
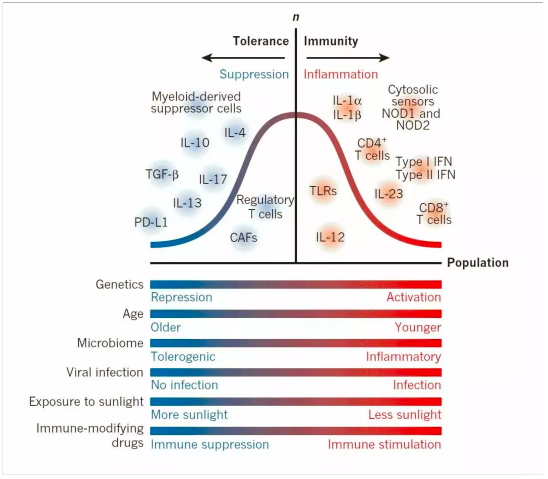
③阐明转移癌细胞离开肿瘤微环境为啥可以存活的机制。理解转移前和转移后微环境的免疫学性质变化。
癌细胞离开创始根据地去宿主的其它地盘上开疆拓土是一件风险极大的事情。进入血液中的癌细胞存活下来的机率非常低,多数被杀死或者自己死亡。能够粘附血管壁并进入周围组织存活下来的又低了许多。不过有证据表明,即使血液中可以检测到存活癌细胞,但最终的解决有以下几种:
-
全挂在血液中或在建设新的根据地的过程中。
-
到了某根据地,但是不适合发展,长期潜伏,甚至终身无用;也有可能,在N年后的某一天,获得合适机会再发展起来。临床上可以观察到很多年后复发的乳腺癌之类。
-
到了根据地,迅速发展起来。
④阐明构建癌症免疫和代谢之间的关系。
代谢是生命的基本特征,所有有生命系统的根本特征。本质上所有的生命都是由基本粒子组成的,基本粒子组成了原子,原子组成了分子,这种不断的系统化后使得系统有了超级复杂性和有序性。为了维持超级系统,系统必须要代谢物质并摄入能量。
免疫是广义的代谢概念的一种,代谢包含了免疫机能在内。多细胞化后的动物代谢负荷加大,为了供应所有的细胞尽可能获得营养物质,多细胞体系特化了循环系统;为了提高摄食效率,发展了运动系统;为了协调运动系统和内在运动系统循环系统的效率,发展了神经系统。神经系统是一种高级精准的协调系统,后来居上地位于内分泌和免疫系统之上,免疫系统是消化系统的补充系统。内分泌系统负责各功能细胞群的远距离近距离通讯,媒介为化学分子。
⑤阐明癌症免疫与细胞行为调控的关系。
人体细胞在过去的35亿年里一直在变化,直至成为今天的细胞形态与功能之前它经历了复杂的演化过程。最近6亿年的演化过程中,人体细胞的祖先是一个开放的系统,接受一切有利于自己的整合。人类基因组中的基因规模目前尚不是很清楚,推测由3万条基因单位。这些基因是长时间和地球外界自然环境相互作用的结果,经历了残酷的自然选择过程和对外界的同化过程,才建立起了今天的规模。人类基因组呈现丰富的多态性,既与演化的分支有关,如6万年前就与欧洲人分开的亚洲人与欧洲人之间存在显著的表型差异;也与外界环境有关,如病毒基因组序列对人类基因组的转染与整合,外界自然有利有害突变的积累,以及生活方式和生殖方式对现有基因组体系构建的影响。
免疫系统的基本架构早在单细胞阶段就已构建了起来。例如免疫球蛋白的结构在人体内是一个巨大的家族,包括了细胞上的受体(TCR)、MHC等,根据FC段的不同又分类为5种免疫球蛋白。到了多细胞阶段后,免疫系统已经分化为一个庞杂巨大的网络系统,虽然经常出错:如不能完全遏制由于人类基因组不稳定性带来的异常增殖性突变,本来细胞的增殖是统一于宏观的神经内分泌调控的。这种增殖可能会伴有移动性病灶扩散,即所谓的恶性增殖、毒性增殖。
题外话:物理学视角下人生演化过程

基本粒子的费米特性,泡利不相容原理,使得原子组成的世界按照层次的原则组建了起来,出现了多层次的生物进化。每个生物个体都是一个宇宙在重演的宇宙的历史,个体人也不例外。多细胞动物人虽然看起来很复杂,其实基本的构成不外代谢与免疫2个,还有一个是与其共生的微生态。
代谢是外在的免疫,免疫是内在的代谢;代谢的本质是同化,免疫的本质是清除。
版权声明
本公众号所有转载文章是出于传递信息,分享热点的目的,同时明确注明来源和作者,不希望被转载的媒体或个人可与我们联系(marketing@Countstar.cn),我们将立即进行删除处理。所有文章仅代表作者观点,不代表本站立场。
